实验四、细菌的系统发育分析
生命就是进化 ——阿米莎·帕泰拉
Life is all about evolution. By Amisha Patel
一.实验目的
- 学习使用Chromos软件或MEGA软件中的分析和处理基因测序峰图
- 学习使用DNAMAN软件拼接基因序列
- 学习利用NCBI网站在线搜索基因序列
- 学习利用MEGA软件构建基因系统发育树
二.实验原理
对测序结果预处理可以采用Chromas软件或者MEGA软件中的TraceEditor插件,它可以用来查看和编辑DNA的峰形图, 图谱文件一般为SCF格式(*.scf)或.abi格式,如下图测序结果所示,高低不一的峰代表基因序列当中的每一个碱基,不同的碱基用不同的颜色表示,从大约第30个碱基开始,碱基峰整齐独立细长,且底线上的杂峰很低,符合分析要求。在测序结果的开始和末尾处的碱基峰杂乱无规则且底峰较高,需要将其剪切掉才能进行序列分析。
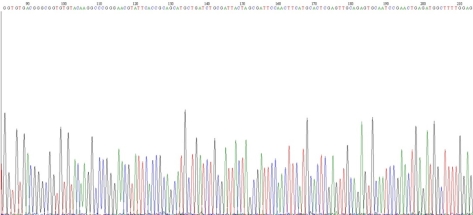
由于16S rRNA的基因较长(1.5kb),测序时要分别从正向和反向进行测序,将测序结果用DNAMAN软件进行拼接,找出两段序列相接处的重复序列,将两段序列拼接为一段连续序列。
得到拼接好的序列后,到NCBI数据库中,使用同源序列比对程序(Blast),寻找与待分析菌株亲缘关系最近的菌株,找出用于构建系统发育树所用的参比菌株和模式菌株的序列(或通过特定网站寻找和下载相关菌株的序列)。
分子系统发生分析主要分成3个步骤,① 分子序列或特征数据的分析;②系统发育树的构建;③结果的检验。系统发育树的构建有很多方法,跟据所处理数据的类型,可以将系统发育树的构建方法大体上分为两类。一类是基于进化距离的构建方法,利用分析单元间的进化距离,依据一定的原则和算法构建系统发育树,如非等重平均连锁(UPGMA)法、邻接(NJ)法、最小进化(ME)法等。另一类方法是基于离散特征的构建方法,如DNA序列中的特定位点核苷酸,建树时着重分析序列间每个特征(如核苷酸位点)的进化关系等,如最大简约(MP)法、最大似然(ML)法等。对基因序列的比对方法、模型选择和建树方法都会影响到系统发育树的拓扑结构,目前最常用的方法为ML法,但速度慢。
在所构建好的系统发育树的每一个分支上都会有一个数字,该数字被叫做当前系统发育树此分支的扩展值(bootstrap)。在建树的过程当中会设置一个重复计算次数,一般ML法建树的重复计算次数设置为100,那么扩展值90就表示在随机重复计算了100次中,此树显示的结果为90次。在一个系统发育树当中,扩展值越高,则结果越可信。
MEGA软件可以用来编辑序列数据、序列比对、构建系统发育树(包括ML法、NJ法等)、推测物种间的进化距离等。此软件的输出结果具有不同形式的可视化效果。
本次实验要分析的是细菌的16S rRNA基因,即16S rDNA序列。16S rRNA是细菌等原核生物核糖体小亚基组成成分中一种RNA,也称为SSU rRNA(small subunit ribosomal RNA),由基因组上16S rDNA编码。16S rRNA或16S rDNA是细菌、古菌等原核生物系统发育分析中最常用的核酸分子。16S rRNA的含量大(约占细菌RNA含量的80%),分子量大小适中,约1.5 kb左右,存在于所有的生物(真核生物为18S rRNA)中,在序列上既有结构与功能上高度保守的区域,又有区分不属的可变区域,素有“细菌化石”之称,常用于确定属及属以上分类单位的亲缘关系。利用16S rRNA恒定区序列特别保守的特点,在恒定区上设计引物,将几乎整个16S rRNA基因扩增出来,得到几乎全长的序列,进行系统发育分析或比较,也可只得扩增出其中一个可变区(全部可变区包括V1-V9),用于比较不同属间的差异。
将所测序结果与GenBank中储存的相关序列进行比对(Blast),计算被分析细菌与已知种类的遗传距离,以确定其系统发育分类地位和分类水平。一般认为16S rRNA或其基因序列分析有助于细菌属以上水平的分类。通常,16S rRNA或16S rDNA序列相似性< 97%,且在进化树上形成稳定的独立分支,又具有特征性碱基,基本可以作为种的界限;而16S rRNA相似性< 95%,且在进化树上形成稳定的独立分支,又具有特征性碱基,基本可以作为属的界限。
三.实验方法
- 软件:Chromas、DNAMAN、MEGA,NCBI-Blast (website)
- 16S rRNA 基因序列
- 计算机
四.实验内容
1. 测序峰图分析和处理
打开Chromas软件,点击左上角的“Open”,
找到想要处理的16S rDNA序列文件,选中后单击“打开”
打开后的测序峰图
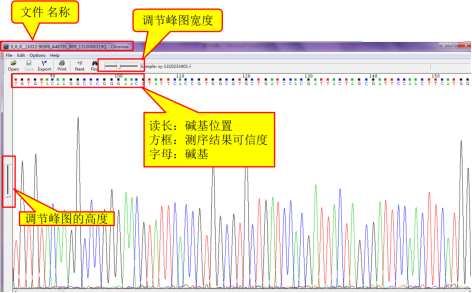
对峰图的首尾进行剪切:首:≥ 30bp (原则是切去有重叠峰的峰段),尾: 视序列质量而定,但剪切后最好不要短于700bp,鼠标点击首部红框位置,即要进行剪切的碱基。

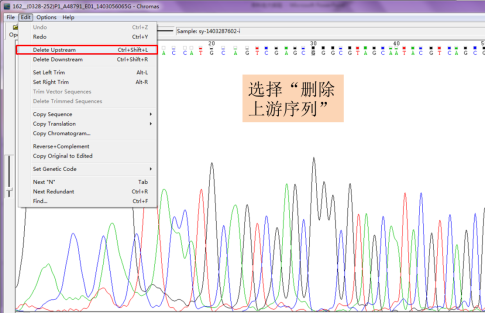
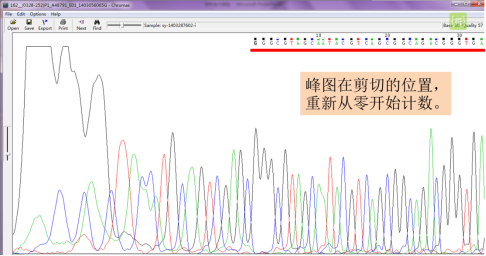
单击“edit”,选择“Delete Upstream”,剪切后的峰图会从剪切的位置重新从零开始计数,
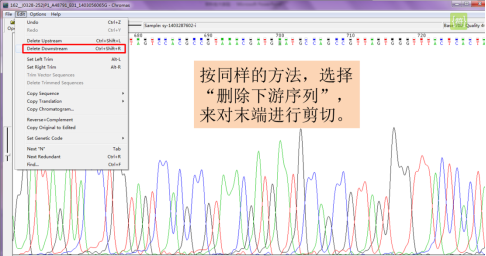
同样的,点击“edit”,选择“Delete Downstream”删除下游序列,
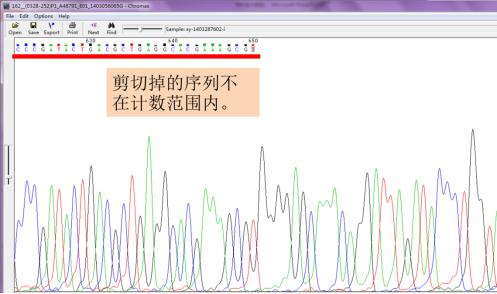
剪切掉的序列将不在计数范围之内,
将剪切后的文件另存为新的文件名,
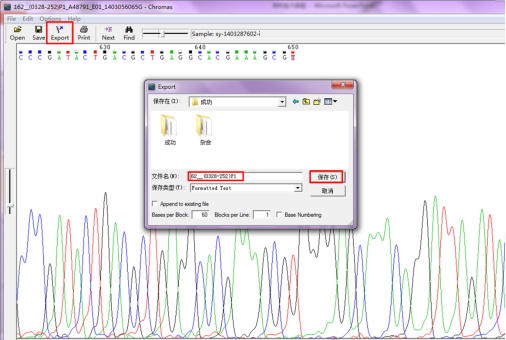
关闭原始测序文件时,不要保存对文件的修改,保留原始文件,以便日后查看。
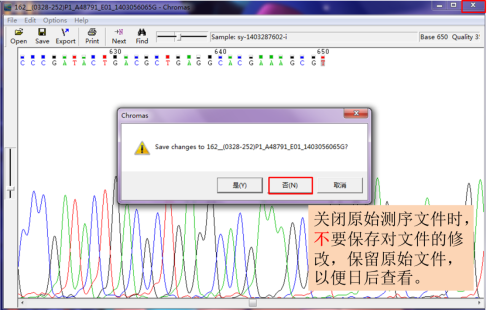
16S rDNA是双向测序,以同样的步骤处理反向序列。
2. 基因序列的拼接
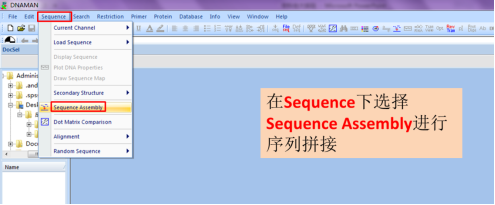
打开DNAman软件,点击“Sequence”,在下拉菜单中选择“Sequence Assembly”进行序列拼接。
点击“Add file”,添加剪切好的正向和反向序列,
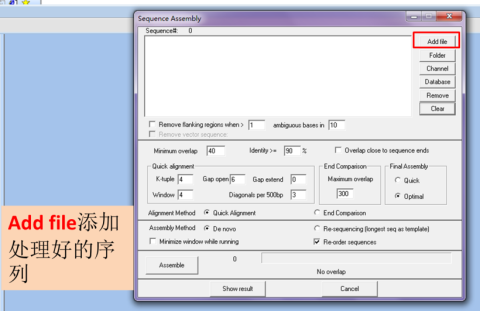
点击“Assemble”,必须有“overlaps”才可继续操作(没有overlap说明在序列剪切的时候,切掉的序列太多,需重新剪切,保留更长的序列),
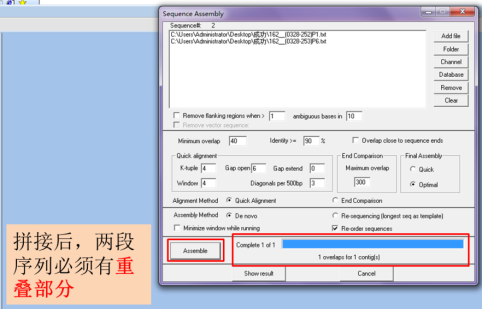
点击“Show result”,
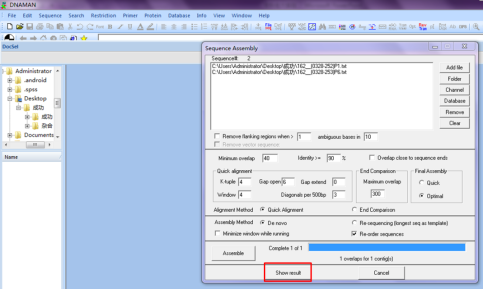
查看输出后的结果,看正向和反向测序结果重叠部分是否一致,不一致时,需要查看原始的峰图修正错误。
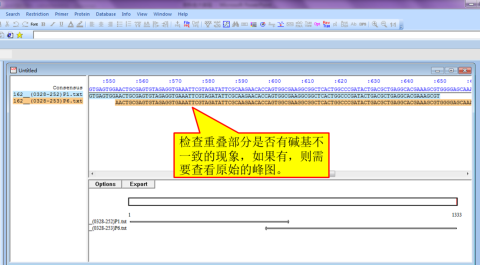
序列检查无误后,点击“Export”,输出序列,
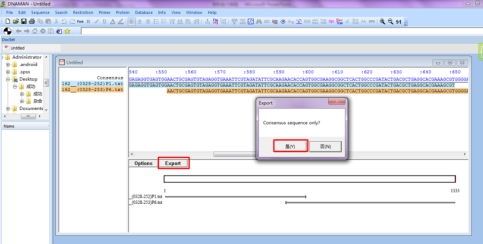
处理输出后的序列,手动删除选中的一段文字,
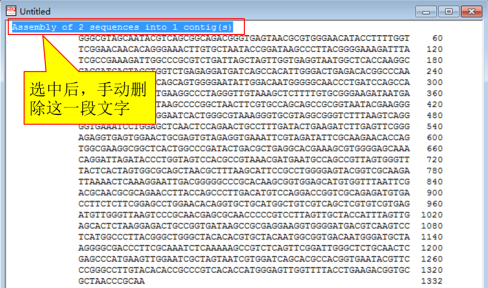
将序列全部选中后,点击右键,选择“Format Sequence”更改序列格式,
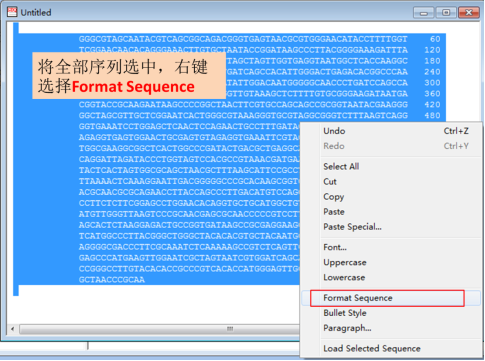
将每行的数字改成900,即最大值,去掉“Number label”前的对勾,
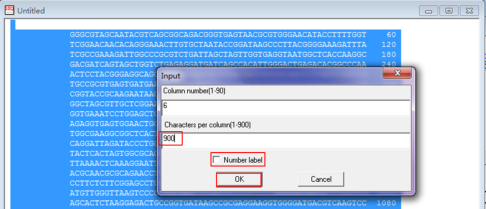
手动删除中间部分的空格,
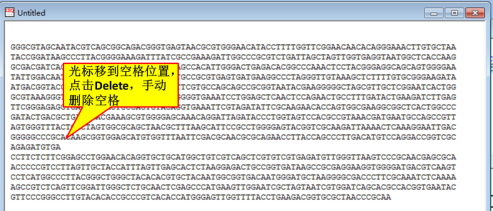
保存处理好后的序列文件, 以用于系统发育树的构建。
3. NCBI-Blast搜索和下载菌株序列
3.1下载已知模式菌株或参比菌株基因序列
打开NCBI网页(http://www.dtd.nlm.nih.gov/),将搜索范围限定为“Nucleotide”, 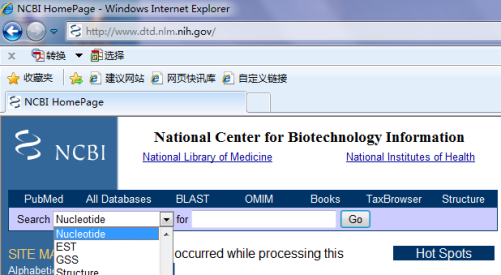
以菌株号做为关键词来搜索菌株的基因序列,点击“Search”,

得到这株菌的全部基因序列, 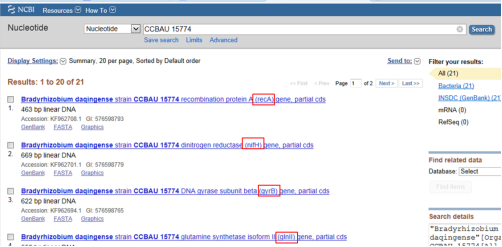
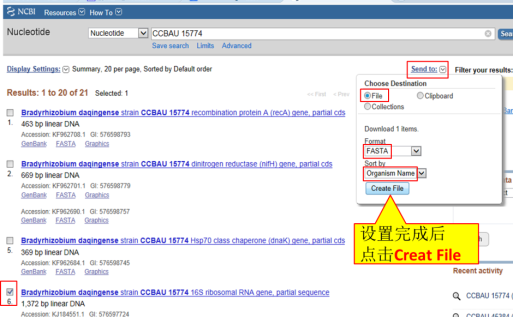
找到16S rDNA序列,单击左侧方框后,显示对勾,右上方点击“Send to – File – FASTA - Organism Name – Create File”即可下载文件;
另外,也可以用菌株某个基因的GenBank号作为关键词进行搜索,

按照相同的方法,把序列下载下来即可,
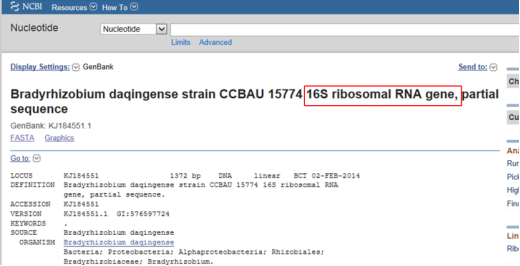
3.2 搜索亲缘关系较近的参比菌株序列
在NCBI网站主页找到Blast选项,
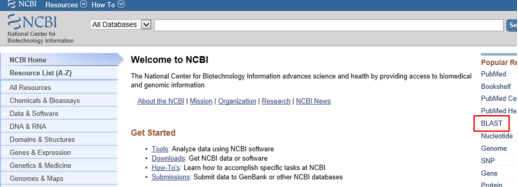
搜索范围选择“nucleotide blast”,
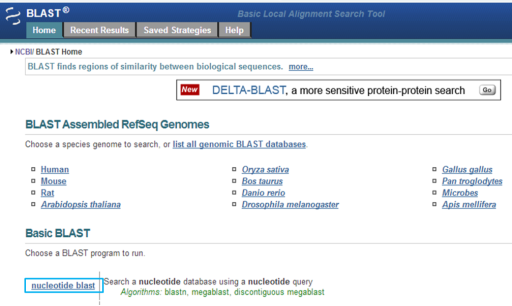
将序列复制到框内—,选择:“Others”—“Highly similar sequences”—“show result on a new window”, 点击“blast”,

根据Blast结果可以知道自己的菌株与哪个属最近,一般处在第一位置的都是相似性最高的菌株,
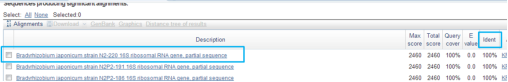
下载序列的第一步是要将菌株前面的方框内勾上对勾,

点击“Download—FASTA—Continue”下载序列文件,可用于序列分析。

4. 16S rRNA基因序列系统发育树的构建
4.1. 基因序列排队和切齐
打开MEGA 5.50软件,点击“Align – Edit/Build Alignment”来新建一个MEGA文件,
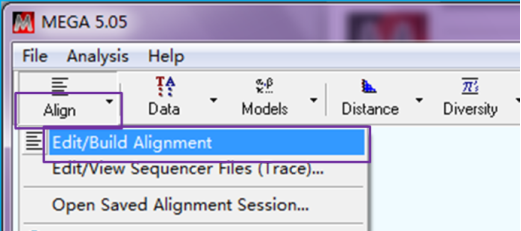
点击“Creat a new alignment - OK”,
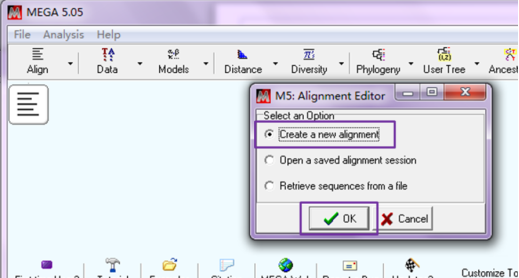
选择“DNA”,
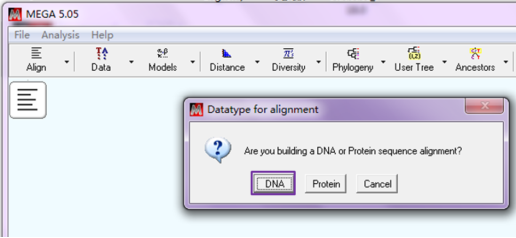
在新建的MEGA文件中点击“Edit—Insert Sequence From File”,从文件中导入序列,
将序列全部导入后,点击“W—Align DNA”,
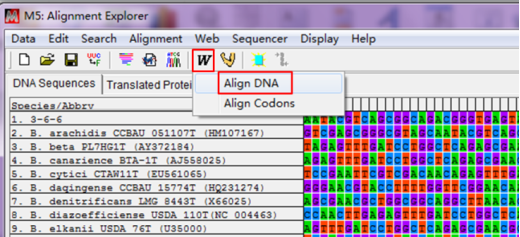
弹出对话框,不用改变弹出的对话框的参数,点击“OK”,
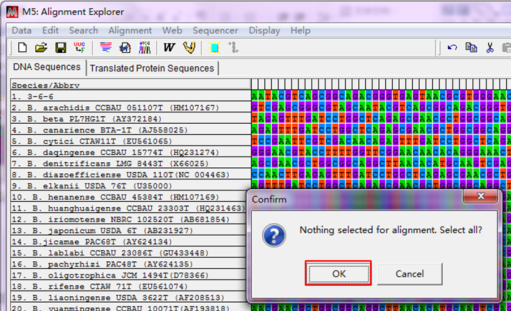
再弹出的对话框,点击“OK”,

排列对齐后,序列大部分是一致的,但也有少数地方有差异,说明这些菌株序列的不同,
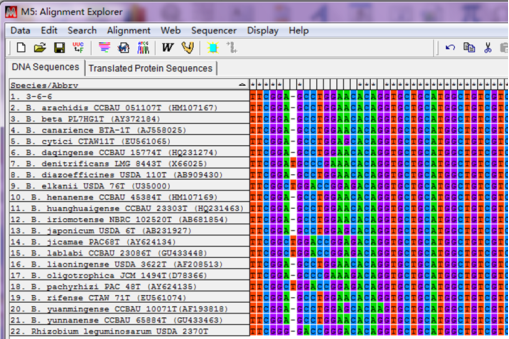
排列对齐后可能遇到的问题,
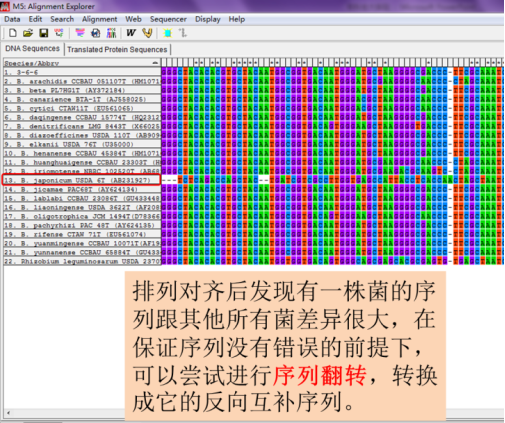
选中序列后,点击“右键—Reverse Complement”对序列进行翻转,
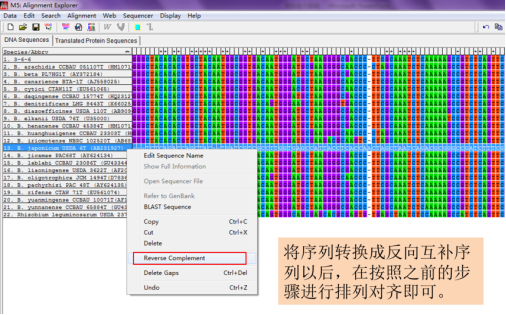
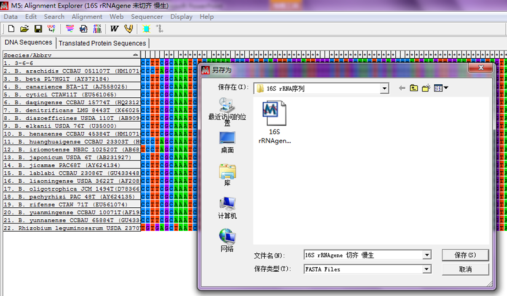
保存比对正确后的序列,选择“Data—Export Alignment—FASTA format”,文件保存时命名通常会加上“未切齐”,
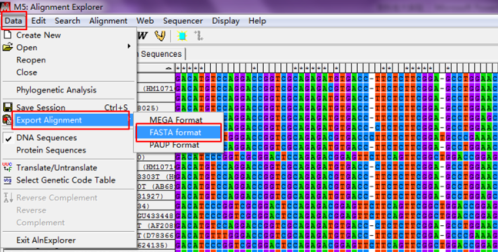
然后对序列进行切齐处理,
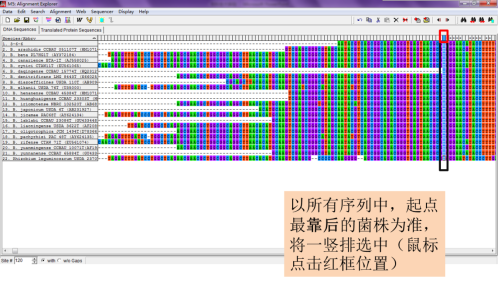

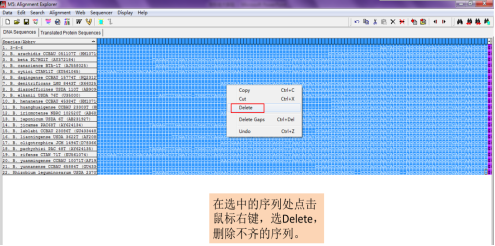
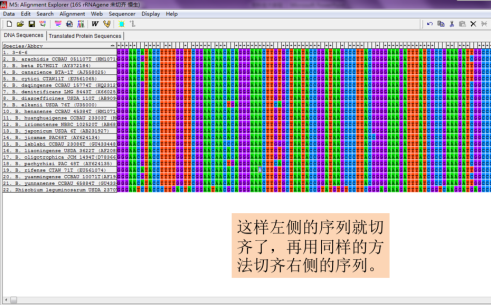
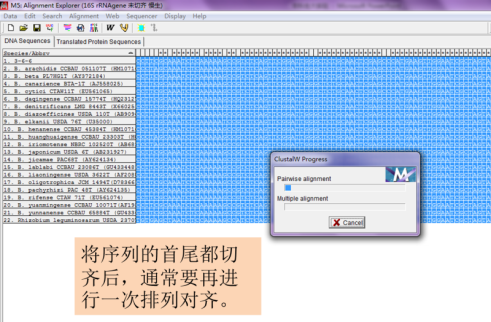
第二次排列对齐后,如出现两边不齐的,再重复一次切齐步骤,然后保存切齐的序列文件,在命名时通常标注“切齐”,
4.2 系统发育树的构建
打开MEGA软件,点击“Phylogeny—Construct/Test Maximum Likelihood Tree ”
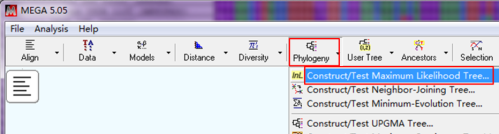
选择切齐的序列文件来构建系统发育树,

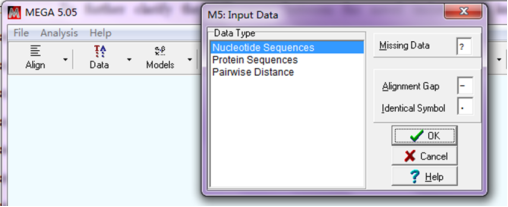
选择“Nucleotide Sequence - OK”,
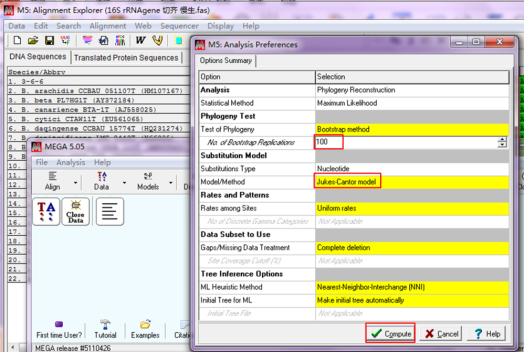
Bootstrap 值为100,建树模型选择“Jukes-Cantor model”,设置好后点击“Compute”
运行完毕后就可以得到16S rDNA 基因的系统发育树(图1)。
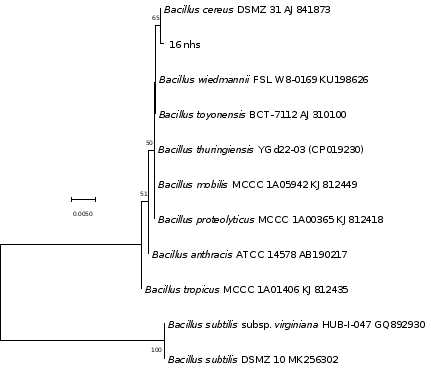
图1 基于16SrDNA的系统发育树
4. 3 菌株间遗传距离的计算
打开MEGA软件,点击“Distance—Computer Pairwise Distances”
参数的设定与之前建树时一致,
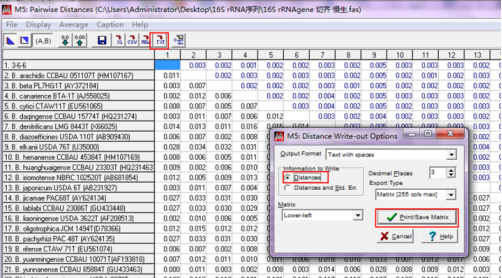
运行完毕后得到菌株间遗传距离的表格,

点击“TXT—Distance—Print/Save Matrix”, 即可将文件以txt. 的格式保存,也可拷屏保存,便于查看。 根据遗传距离可以计算出任何两个菌株之间的序列相似性(序列相似性=1-遗传距离)。
五.作业与思考题
1. 说明16S rRNA序列分析在系统细菌学中的重要意义。
2. 16S rRNA上的可变区、保守区有哪些?
3. 做出16S rDNA系统发育树聚类图 (附图)。
4. 计算测序菌株与参比菌株的序列相似性(附表)。
5. 根据系统发育树,分析测序菌株的系统发育关系和分类地位。
6. 查阅文献,或查阅你分离到的细菌的基因组,或与之最近的菌的基因组,分析你分离到的这株菌有无分解分解纤维素的功能,或者有无相关基因?
六.拓展学习
16S rRNA的二级结构分析https://rnacentral.org/rna/URS0000381BA4/424182?tab=2d、可变区、保守区分析。
七.参考文献
1. 隋新华主编《原核生物进化与系统分类学实验教程》,科学出版社,北京,2016年01月。
八. 相关术语或关键词
16S rRNA 16S rDNA Chromas软件 MEGA软件 DNAman NCBI网站
Blast 序列相似性 序列拼接 序列比对 FASTA格式序列
九. 问题反馈
对于本实验指导的内容,如有问题、疑问或修改建议,请致信:chenwf@cau.edu.cn。编写者将认真对待你的来信并给予及时回复。在此先表示感谢。